

What is TDT? Transfusion-dependent beta (β)-thalassemia (TDT) is a severe genetic disease that results in reduced or absent production of functional beta-globin1,2
The incidence and prevalence of beta-thalassemia vary around the world3
Beta-thalassemia is primarily found in South Asia, the Middle East, North Africa, and Southern Europe. While migration is changing the global distribution of the disease, beta-thalassemia is still considered to be a rare disease in the US and most of Europe, and the exact prevalence of TDT in the US is not known.1,3-5
About 1.5% of the global population (80-90 million people) are carriers of beta-thalassemia. 60,000 symptomatic individuals are born annually.3,5
TDT greatly limits or reduces a person’s ability to produce HbA3,6
Beta-thalassemia is an autosomal recessive disease caused by a mutation in or near the HBB gene that results in reduced or absent production of the beta-globin protein.2,7 Over 350 disease-causing genetic mutations have been identified, most of which are point mutations.8
Deficient beta-globin synthesis impairs HbA production1
Adult hemoglobin (HbA) is a tetramer that is made up of 2 alpha (α)-globin subunits and 2 beta (β)-globin subunits. The number of beta-globins must precisely match that of alpha-globins. If not, the alpha-/beta-globin imbalance impairs the body’s ability to produce functional HbA.1,2,9
NORMAL HEMOGLOBIN

(BETA-THALASSEMIA MAJOR)

*Faded blue indicates reduced or absent beta-globin.
In the absence of sufficient beta-globin, excess unpaired alpha-globin impairs the development and survival of red blood cells (RBCs), leading to chronic anemia and other serious complications.2
Deficient beta-globin synthesis is a defining characteristic of beta-thalassemia2
Deficient beta-globin synthesis is a defining characteristic of beta-thalassemia2
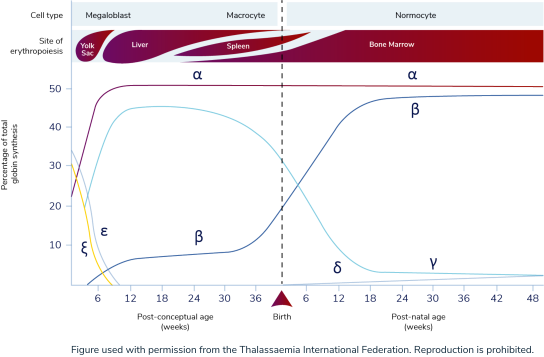
HbA becomes the primary form of hemoglobin after 6 months of age1,9
Under normal conditions, adult hemoglobin (HbA) becomes the predominant form of hemoglobin (Hb) in the body by about 18 to 36 weeks of age, accounting for ≥90% of total Hb.1,9 Prior to HbA production, the predominant form of hemoglobin is fetal hemoglobin (HbF), which is composed of 2 alpha (α)-globin subunits and 2 gamma (γ)-globin subunits.9
Stages of globin protein synthesis1
Stages of globin protein synthesis1
HbA has a higher binding affinity for 2,3-DPG than HbF to allow optimal oxygen delivery to the body10
HbF is important for oxygen transport during fetal gestation because it has a high affinity for oxygen and a decreased affinity for 2,3-diphosphoglyceric acid (2,3-DPG) relative to HbA.10 In spite of HbF’s high oxygen affinity, the low oxygen tension in the fetus allows for proper unloading of oxygen.11 This allows HbF to easily bind to oxygen found in the maternal bloodstream. However, post-birth, HbA is more optimal because it has a higher affinity for 2,3-DPG, which enables oxygen release; this is essential for RBCs to deliver oxygen to the organs and tissues of the body. The switch from HbF to HbA at the appropriate time in an infant’s life is critical to the normal functioning of the body as it grows.10
Adults who express variants of Hb with high oxygen affinity (including HbF) have been shown to deliver less oxygen than normal to tissues, leading to mild tissue hypoxia and, over time, to polycythemia and thrombotic events12
Adults who express variants of Hb with high oxygen affinity (including HbF) have been shown to deliver less oxygen than normal to tissues, leading to mild tissue hypoxia and, over time, to polycythemia and thrombotic events12
Without sufficient beta-globin, excess alpha-globin protein can lead to ineffective erythropoiesis and hemolysis1,2,13
When beta-globin is absent, alpha-globin and its degradation products precipitate, causing ineffective erythropoiesis and hemolysis, which lead to anemia.1,3
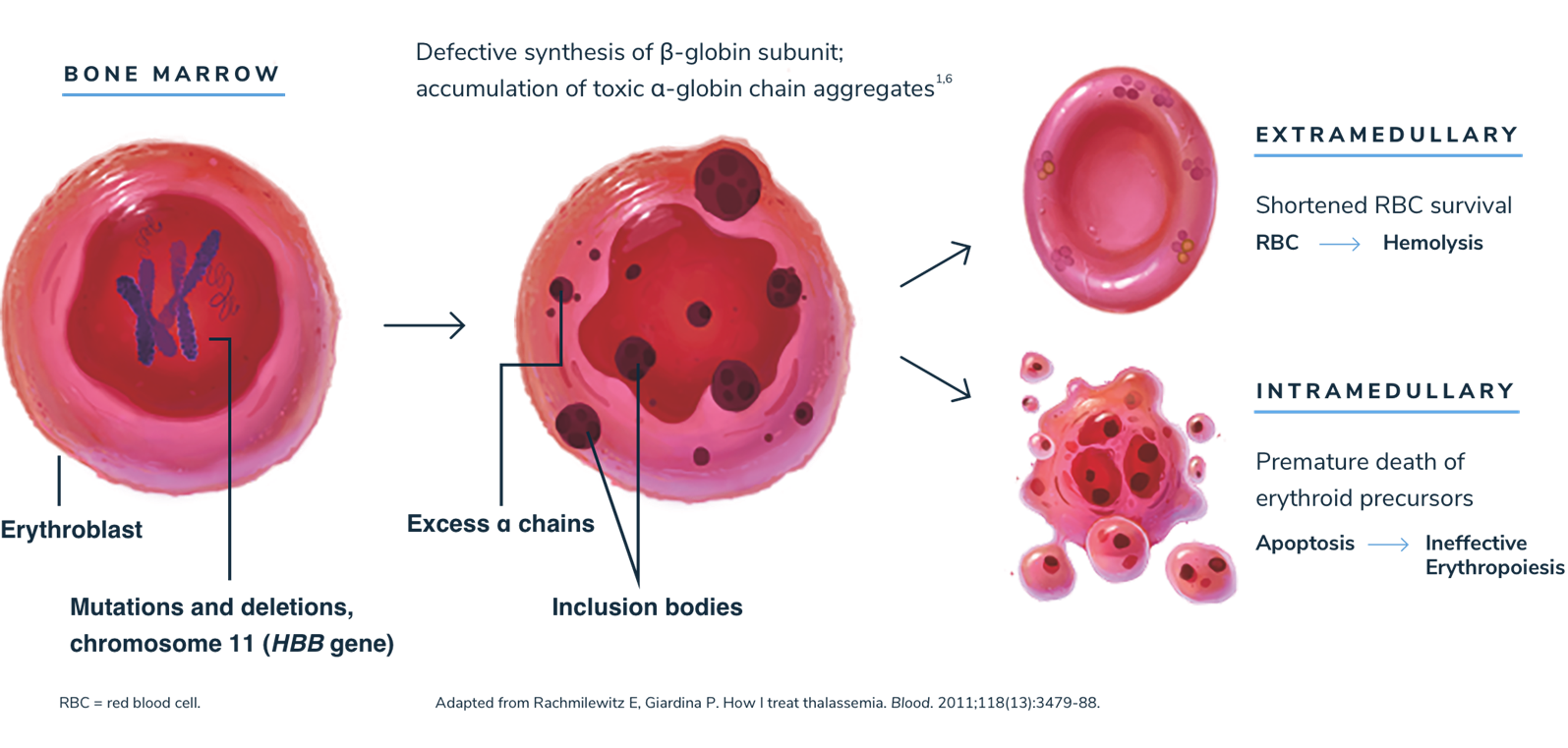
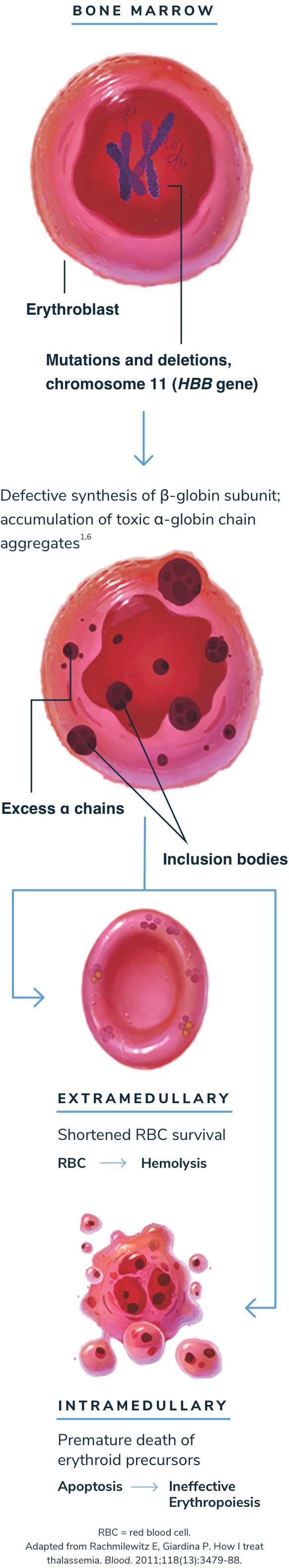
Anemia stimulates erythropoietin synthesis, resulting in intense proliferation of the bone marrow, skeletal deformities, and a variety of growth and metabolic abnormalities. Splenomegaly is typically seen in patients with beta-thalassemia as a result of extramedullary hematopoiesis or as a response to extravascular hemolysis.1,3
The classification of beta-thalassemia is evolving1,2,7
The 2021 Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT), published by the Thalassaemia International Federation (TIF), classify beta-thalassemia phenotypically into two main groups based on clinical severity and transfusion requirement1:
Historically, beta-thalassemia has been classified into three groups: beta-thalassemia minor (trait), intermedia, and major.1,7 These terms may still be used by patients and some clinicians today.
Beta-thalassemia major usually presents clinically between the ages of 6 and 24 months1
Screening for beta-thalassemia can be done by using hemoglobin electrophoresis, high-pressure liquid chromatography, or beta‑globin gene sequencing. Affected infants1:
- have severe microcytic anemia
- fail to thrive
- become progressively pale
- develop hepatosplenomegaly that may distend the abdomen
- have mild jaundice
- may have feeding problems and recurrent fevers due to hypermetabolic state or intercurrent infection
Beta-thalassemia intermedia usually presents at a later age with a milder form of these clinical findings. Those on the more severe end of the spectrum may show slow development and retarded growth, while those on the mild end may be completely asymptomatic, with just mild anemia. People with beta-thalassemia carrier state (heterozygous) show no important clinical effects since the activity of their normal beta-globin gene makes enough stable globin.1
Visit LifeWithBetaThal.com for resources that may help your patients better understand the genetics behind TDT, managing their condition, and how they can plan for their future.

Actor portrayal. Not a real patient.
Take the Beta-Thalassemia
Challenge


Register to receive patient resources, additional education about beta-thalassemia, and updates from bluebird bio.