

Transfusion Therapy for TDT For patients with transfusion-dependent beta (β)-thalassemia (TDT), lifelong transfusions provide symptom management1
Lifelong supportive care with regular RBC transfusions is necessary in TDT1
Red blood cell (RBC) transfusions, which are central to the treatment of severe disease, enable survival by helping to correct the anemia characteristic of beta-thalassemia and limit bone marrow expansion. However, many challenges, such as organ damage due to underlying disease and iron overload, still exist.1,2
Transfusions address Hb levels temporarily1
Male
2-9 years 11.5-14.5 g/dL
2-9 years 11.5-14.5 g/dL
10-17 years 12.5-16.1 g/dL
10-17 years 12.5-16.1 g/dL
≥18 years 13.5-18 g/dL
Female
2-9 years 11.5-14.5 g/dL
2-9 years 11.5-14.5 g/dL
10-17 years 12.5-15 g/dL
10-17 years 12.5-15 g/dL
≥18 years 12.5-16 g/dL
The 2021 Guidelines for the Management of TDT, published by the Thalassaemia International Federation (TIF), recommend regular lifelong transfusions, typically every 2 to 5 weeks, in patients who meet the following criteria1:
- Confirmed diagnosis of beta-thalassemia
- Laboratory criteria
- Hb level <7.0 g/dL on 2 occasions, >2 weeks apart (excluding all other contributory causes such as infections)
AND/OR
- Confirmed diagnosis of beta-thalassemia
- Laboratory criteria
- Hb level <7.0 g/dL on 2 occasions,
>2 weeks apart (excluding all other
contributory causes such as infections)
- Hb level <7.0 g/dL on 2 occasions,
AND/OR
- Clinical criteria irrespective of hemoglobin level:
- Significant symptoms of anemia
- Poor growth/failure to thrive
- Complications from excessive intramedullary hematopoiesis, such as pathological fracture and facial changes
- Clinically significant extramedullary hematopoiesis
The Thalassaemia International Federation (TIF)1,4
Founded in 1987 by a small group of patients and parents, TIF is a nonprofit, nongovernmental organization dedicated to promoting appropriate clinical management of thalassemia in every affected country.4
Since 1996, TIF has had official relations with the World Health Organization (WHO). The outcome of this partnership is an extensive network of collaboration between scientific and medical professionals from more than 60 countries around the world. To further its mission, TIF has published important clinical documents for thalassemia, including guidelines for managing and caring for patients with both TDT and non–transfusion-dependent beta-thalassemia.1,4
The above content was used with the approval of TIF. bluebird bio does not endorse the nonprofit organization or any of its affiliates.
It is essential to maintain hemoglobin levels in patients with TDT1,5
The TIF guidelines recommend regular blood transfusions, usually administered every 2 to 5 weeks, to maintain a pretransfusion level of 9.5 to 10.5 g/dL, which has been shown to help1:
- promote normal growth1
- allow normal physical activities1
- adequately suppress bone marrow activity1
- improve anemia6
- minimize transfusional iron accumulation and ineffective erythropoiesis1,6
- avoid skeletal abnormalities, organomegaly, and growth retardation in children1,6
Because hemoglobin levels are higher post-transfusion, which increases the risk of stroke, post-transfusion hemoglobin target should be 13 to 15 g/dL.1
TIF Guidelines for Transfusion: Maintaining a Pretransfusion Level of 9.5 to 10.5 g/dL1*
 SWIPE OR ROTATE
SWIPE OR ROTATE
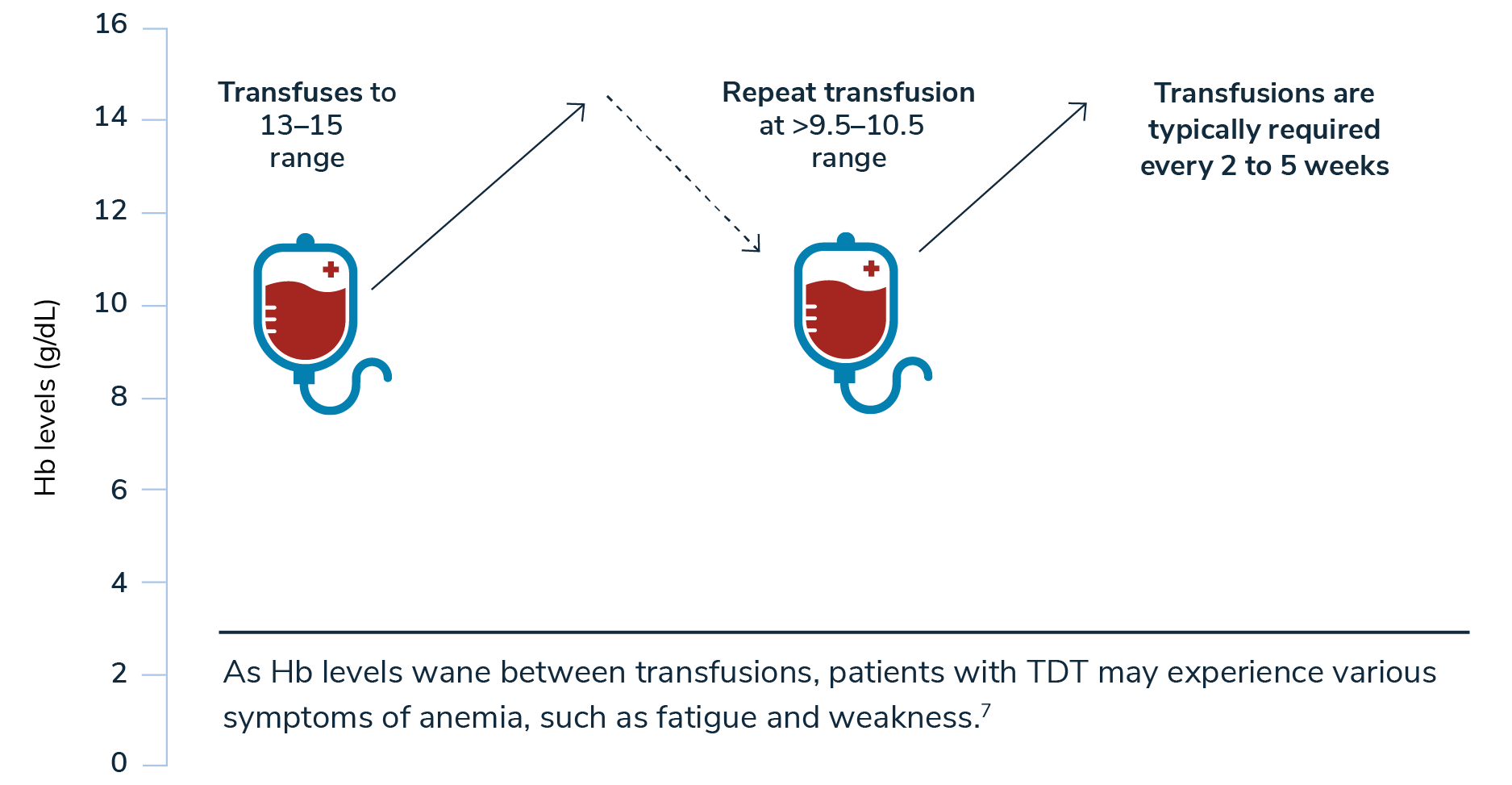
*A higher target pretransfusion hemoglobin level of 11 to 12 g/dL may be appropriate for certain patients with heart disease or other comorbidities.1
Hemoglobin levels should be measured intermittently after transfusion to determine how quickly hemoglobin levels drop. This decrease may be valuable for evaluating the effects of changes in the transfusion regimen.1
Understanding your patient’s symptoms between transfusions may
help you optimize their treatment schedule.
help you optimize their treatment schedule.
As hemoglobin levels wane between transfusions, patients with TDT may experience various symptoms of anemia, such as1,6,7:
- fatigue
- weakness
Patients may also experience transfusion-associated complications, such as1:
- transfusion reactions
- alloimmunization
- infections
Over the years, a greater understanding of the pathophysiology of beta-thalassemia has helped improve disease management and drive the investigation of future potential therapies.1 As an example, today adult patients with beta-thalassemia receiving regular transfusions may have the option of adding an erythroid maturation agent to their chronic transfusion schedule to help moderate their transfusions.1
Regular transfusions enable survival but only temporarily address the symptoms of TDT.1
Actor portrayal. Not a real patient.
Take the Beta-Thalassemia
Challenge
Actor portrayal. Not a real patient.

Register to receive patient resources, additional education about beta-thalassemia, and updates from bluebird bio.